HEMOSTAZA
Hemostaza
este un ansamblu de fenomene care au loc ca raspuns la leziunea unui vas
si care au drept efect oprirea sangerarii.
La
acest proces participa factori vasculari, trombocitari si plasmatici
avand ca rezultat formarea fibrinei. Procesul este contrabalansat de
activitatea sistemului fibrinolitic.
Se
distinge o hemostaza primara asigurata prin interactiunea
intre peretele vascular si placutele sanguine si o
hemostaza secundara care implica formarea de fibrina.
Fazele hemostazei:
faza vasculara;
faza plachetara;
faza de activare a factorilor
coagularii;
fibrinoliza;
activarea sistemului monocit
macrofag care capteaza monomerii de fibrina, produsii de
degradare ai fibrinei si factorii coagularii in exces,
faza reologica in care
fluxul sanguin spala locul leziunilor endoteliale, reduc numarul de
trombocite, scad factorii coagularii si ai fibrinolizei;
faza de actiune a anticoagulantilor
naturali care finalizeaza hemostaza.
1.
Hemostaza primara
Hemostaza
primara dureaza 2- 4 minute si realizeaza oprirea
temporara a sangerarii prin formarea unui trombus plachetar - cheagul
alb - ca rezultat al mecanismelor vasculare si a trombocitelor. Se
numeste si faza vasculo plachetara.
I) Rolul peretelui vascular in hemostaza
a).
Rolul vasoconstrictiei
Vasoconstrictia
postlezionala se datoreaza contractiei celulelor musculare
netede(reflex de axon) si este o proprietate intrinseca a vaselor
mici in special arteriale si sfincterele precapilare: capilarele nu
contin fibre musculare si deci nu sunt contractile; arteriolele
si sfincterele precapilare in leziunile mici pot asigura singure hemostaza; suportul
biochimic este asigurat de catecolamine, serotonine, tromboxanul A2, bradikinina, ce
intervin pe rand in procesul de hemostaza si efectul lor se poate
suma.
b).
Rolul endoteliilor vasculare(EV)
EV
sunt reprezentate de un strat monocelular aflat la interfata dintre sange
si structurile subiacente. Functia CE este de bariera,
secretorie si antigenica de suprafata.
In
conditii normale E.V. sintetizeaza si secreta: factor Von
Willebrand cu rol in aderarea placutelor sanguine; factor V
procoagulant; inhibitor al activarii plasminogenului (PAI), factori
procoagulanti. Dar, predomina mecanismele anticoagulante si
antitrombotice.
In
conditii patologice cand endoteliile sunt lezate sub actiunea
endotoxinei, a radicalilor superoxizi din leucocite si in urma
stimularii cu citokine proinflamatorii ca IL - 1 si TNF (factor de
necroza a tumorilor) se produce:
accelerarea eliberarii de
factor Von Willebrand din endotelii;
expunere de factor tisular
(tromboplastina) la suprafata celulelor endoteliale (CE);
creste productia de
inhibitor al activarii plasminogenului (PAI) care reduce fibrinoliza;
endoteliile expun o proteina
care favorizeaza aderarea leucocitelor si diapedeza;
sub actiunea trombinei
endoteliile produc si elibereaza factor de activare a
placutelor(PAF) care activeaza local placutele si amplifica
procesul de aderare.
Toate
aceste modificari favorizeaza formarea dopului hemostatic prin
aderarea de placute la peretele vascular (mediat de f. Von
Willebrand), prin initierea fibrinoformarii pe cale extrinseca
si prin limitarea fibrinolizei.
c)
Rolul celulelor subendoteliale
Fibrele
de colagen, microfibrilele de elastina, membrana bazala
reprezinta principalele elemente vasculare la care are loc aderarea
placutelor si initierea hemostazei.
Interactiunea
dintre colagen si placutele sanguine poate fi perturbata
din cauza unor anomalii ale placutelor sau a unei structuri anormale
a colagenului.
d) Rolul celulelor musculare netede
Celulelor
musculare netede cu exceptia capilarelor sunt prezente in tot arborele
vascular mai ales in arterele de calibru mic.
Au
functie: contractila - ingusteaza lumenul vaselor lezate; sintetizeaza
colagen, proteoglicani si proteine din structura fibrelor elastice; au rol
in procesele de separare a peretelui vascular lezat.
In
leziuni ale vaselor mici(arteriale, capilare, venale) prin vasoconstrictie, aderarea placutelor sanguine la
subendoteliu si agregarea placutelor se poate limita fluxul
sanguin, dopul hemostatic consolidandu-se prin fibrinoformare.
In hemoragiile pe artere de tip elastic sau
vene de calibru mare nu pot fi oprite prin mecanisme proprii; necesita
oprirea pe cale chirurgicala.
Vasculopatiile
Afectiunile
peretelui vascular sunt luate in considerare atunci cand se exclud deficitelor
placutelor sanguine si cand coagularea este normala.
Se
pot prezenta ca:
angiectazii, dilatatii
vasculare, in care sangele ramane in interiorul vasului;
purpure in care sangele este
extravazat in tesuturi, in special in teritoriul cutanat; nu dispar
dupa vitropresiune (presiune cu o lama de sticla).
Purpura
vasculara este formata din leziuni punctiforme (sub 3 mm) denumite
petesii, localizate mai ales la membrele inferioare unde sunt supuse la un
maxim de presiune venoasa retrograda. In purpurele vasculare cu caracter inflamator (vasculite) leziunile
sunt usor elevate fiind palpabile.
Hematiile
extravazate pot fi indepartate din piele intrand in limfatice sau pot fi
degradate in macrofage unde nucleul porfirinic al Hb, se metabolizeaza
spre bilirubina (coloratia din rosie in galben) in timp
de Fe-ul se depune sub forma de hemosiderina. In formele cronice
hemosiderina acumulata determina coloratia bruna a
tegumentelor.
Vasopatii ereditare
1. Teleangectazia hemoragica
ereditara = b. Rendu - Weber -
Osler este o anomalie ereditara ce se
transmite printr-un mecanism autosomal dominant, afecteaza ambele sexe;
este o dilatare patologica a vaselor terminale in special venele prin
defect de angiogeneza a vaselor mici care au traiect serpuitor, sunt
ectaziate, fragile( sunt formate doar din endoteliu fara tunica
si conjunctiva); devin mai palide sau dispar la vitropresiune;
isi pastreaza tot timpul aceeasi culoare.
Diagnosticul se
stabileste prin triada: hemoragii recurente; teleangectazii multiple;
antecedente heredocolaterale.
2. Boala
FABRY - angiokeratoma corporis diffusum. Este o tezaurismoza cu sfingolipide ce se depun in piele, vasele
mici din creier, rinichi, inima si leziuni nodulare in scrot,
periombilical si fese. Are un mecanism de transmitere legat de cromozomul
X si se manifesta deci la sexul masculin.
3. Ataxia - teleangiectazica
Este
o boala ereditara cu transmitere autosomal recisiva. Se
asociaza cu ataxia, deficite imune, endocrine, intelectuale.
4. Hemangiomul cavernos - sindrom Kasabach -
Merrit
Este o malformatie
vasculara cu aspect de zmeura insotita de modificari
si-n vasele din splina sau ficat.
Anomalii ereditare
ale tesutului conjunctiv.
Hemoragiile
se produc datorita reducere rolului de suport al structurilor
perivasculare(colagen, elastina) ce sustin zona endo si
subendoteliala avand drept consecinta o fragilitate
crescuta a vaselor(sangerari).
1. Sindrom Ehlers - Danlos (9 tipuri de anomalii ale colagenului): articulatii hipermobile;
tesuturi fragile; sangerari spontane sau la traume mici( echimoze
intinse si hematoame subcutanate).
2. Pseudoxantomul elastic este o boala cu transmitere autosomal recisiva, un deficit
congenital de hidroxilizina ce afecteaza fibra elastica din
arterele mici(fibrele elastice din piele si media arterelor este
anormala); teleangiectazii in piele, echimoze; sangerari in creier,
retina, tub digestiv, urina, uter.
3. Osteogeneza imperfecta, boala cu transmitere autosomal dominanta caracterizata
prin fracturi, diformitati prin deficit de formare a matricei osoase.
4. Sindromul Marfan este
o anomalie cu transmitere autosomal dominanta ce
consta intr-un deficit de legare transversala a fibrelor de colagen
caracterizata prin: extremitati foarte lungi(om paianjen);
dislocarea cristalinului. Se poate insoti de: insuficienta
aortica; anevrism disecant; risc de hemoragii; echimoze dupa
traumatisme minime.
Aproape
toate anomaliile tesutului conjunctiv se insotesc de anomalii ale
placutelor sanguine care sunt hiporeactive, mai ales in secretia
plachetara.
Vasopatii
castigate
Privesc doar afectiunile vasculare care
evolueaza cu anomalii ale hemostazei evidentiabile prin petesii
sau echimoze.
1. Purpure mecanice
prin: traumatisme, suctiuni sau muscaturi; cresteri ale
presiunii venoase retrograde: cresterea presiunii in vena cava
superioara in tumori toracice, accese de tuse, voma ce duc la
petesii de fata si gat; purpura ortostatica pe
membrele inferioare prin jartiere sau varice venoase.
2. Purpure prin atrofia tesutului de
sustinere.
a) Purpura senila este urmare a scaderii
progresive a colagenului din piele si peretele vascular. Clinic: leziuni
echimotice pe fata extensoare a bratelor, marginea radiala a
antebratelor, fata dorsala a mainilor si gatului; semnul
ochelarilor la baza nasului; prin incarcarea cu Fe a macrofagelor
echimozele raman maronii si nu se trateaza. Hipermobilitatea
pielii pe tesuturile subjacente favorizeaza lezarea vaselor din derm
si aparitia de echimoze.
b) Scorbutul, avitaminoza
C este cauzata de deficitul sever de vit. C care scade activitatea enzimei
prolinhidroxilaza(esentiala in sinteza colagenului), reducand
procesul de hidroxilare a prolinei si lizinei. Precursorii lor intra
in structura colagenului si ii stabilizeaza structura.
Rezultatul
este o scadere cantitativa si calitativa a colagenului
si fragilizarea vaselor de calibru mic.
Clinic:
hemoragii gingivale, musculare, tesut celular subcutanat petesii pe
fata interna a coapselor, fese, fata mediana a
bratelor(in jurul foliculului de par); la copii hemoragii
subperiostale; hiperkeratoza a pielii; gingii tumefiate si dureroase,
pierderea dintilor datorita afectarii colagenului din ligamentul
alveolo dentar.
Paraclinic: anemie
moderata, trombopenie pasagera, semnul garoului este pozitiv, timpul
de sangerare este prelungit. Doze de 25 mg/zi de vit. C o vindeca.
c) Excesul de corticoizi prin exagerare a secretiei endogene in sindromul Cushing si
terapie prelungita.
Mecanism:
reducerea sintezei de colagen; hipofunctia macrofagelor.
3. Purpure infectioase prin leziuni la nivelul CE, apar in boli infectioase de natura:
bacteriana: scarlatina,
febra tifoida, meningita, dizenterie, septicemii, endocardite;
ricketsiana: tifos
exantematic, febra patata a Muntilor Stancosi,
virotica: rugeola,
variola, gripa; protozoare: malaria;
Mecanisme:
lezarea directa a peretelui vascular de catre microorganism-
ricketsioze sau directa a peretelui de catre toxine: septicemii cu
germeni gram-negativi.
4. Vasculitele
Se
refera la un proces inflamator al vaselor sanguine. In general se
datoreaza unor perturbari ale raspunsului imun constituind vasculitele
prin hipersensibilizare.
Patogeneza
lor este legata de formarea de C.I. local sau in circulatie.
Factori
etiologici: produse sanguine ca in boala serului, medicamente: antibiotice,
antiinflamatoare, diuretice, barbiturice, infectii: streptococice,
hepatita B, lepra, tbc, herpes, rubeola, HIV, neoplazii: carcinoame,
limfoame, leucemii, colagenoze:LED, P.N., P.R.
a) Purpura Hipergamaglobulinemica
Waldenstrom: cresterea policlonala a
gamaglobulinelor; purpura, valuri de eruptie petesiala pe
membrele inferioare, pimentatie bruna pe gambe(dg. retroactiv
dupa 3-5 ani) cresterea V.S.H., vascozitate crescuta a serului,
C.I. circulante, crioglobulinemice, hemoragii retiniene, alterarea nervilor
periferici ce determina fie polinevrita senzitivo motorie sau
necroza tegumentara.
Mecanisme:
vasculita, hipervascozitate; modificari ale placutelor
sanguine si ale coagularii.
b) Purpura Henoch - Schonlein(
purpura alergica), este o purpura
vasculara distincta, autoliminanta ce apare la copii si
afecteaza pielea, rinichii, intestinul si articulatiile. Este
precedata cu 1 - 3 saptamani de o stare febrila, IACRS,
infectii streptococice, alergii alimentare sau medicamentoase. Mecanismul
este imunologic printr-o reactie de hipersensibilizare de tip III cu
formarea de complexe imune ce contin Ag streptococic in exces. Este
insotita de fenomene acute cu ascensiune febrila: dureri
articulare, purpura cutanata: petesii cu urticarie, papule,
edem, forme de necroza cutanata; colici abdominale cu scaune
muco-sanguinolente; infuziuni sanguine in rinichi, testicule, peritoneu,
pericard, glob ocular; melena , perforatii intestinale, abdomen acut
sau fals abdomen acut(colici periombilicale); renal(hematurie, GNA focala
de tip mezangial, proteinurie, edeme); cresterea IgA in ser, depozite de
IgA in piele, rinichi; teste de hemostaza normale.
Rolul trombocitului in
hemostaza
Placutele
sanguine sunt fragmente celulare lipsite de nucleu. Obisnuit, circula
sub forma de discuri biconvexe cu suprafata neteda. In
leziunile vasculare adera la tesuturile subendoteliale, devin
globuloase, se strang si formeaza gramezi (agregate)
varsandu-si o parte din continutul unor granule. Numar 150
- 350000 /mm3 ;durata de
viata 8 - 11 zile.
Aderarea trombocitara
la nivelul leziunii are la baza un proces fizic de natura
electrostatica: CE lezate se incarca pozitiv si vor atrage
trombocitele incarcate negativ.
Activarea plachetelor se face de catre
stimuli:
o fiziologici: trombina, colagen, ADP, adrenalina,
o patologici: complexe Ag - Ac; gamaglobuline agregate; virusuri; PAF -
factor activator al placutelor produs de leucocitele activate;
In
procesul de activare se genereaza produsi de metabolism ai acidului
arahidonic - tromboxan A2 care amplifica raspunsul
plachetar la stimuli.
Aderarea este continuata
prin ancorarea plachetelor la fibrele de colagen din zona endoteliala
si punerea lor in contact cu f v.W prin intermediul unor receptori
specifici, glicoproteine Ib.
In continuare adezivitatea trombocitara
va fi amplificata prin doua mecanisme:
fibronectinul secretat de CE se ataseaza de f vW si-i
creste capacitatea de aderare le trombocit;
fibrinogenul se ataseaza de trombocit la nivelul GPIIb si
IIIa formand punti macromoleculare intre trombocite.
Urmeaza agregarea
trombocitelor desfasurata in doua valuri sub
influenta:
ADP din CE si fibrele de colagen(h.agregant);
Al 2 lea val este declansat de TXA2 din fosfolipidele
membranare degradate de catre ciclooxigenaza.
TXA2
determina contractia citoscheletului trombocitar si
contractia fibrelor musculare vasculare. Membrana trombocitara devine
neregulata, cu spicului care favorizeaza agregarea celulelor intre
ele, isi pierd forma si secreta intracelular: Ca, F 3 si 4
plachetar, serotonina(vasoconstrictie), factor de crestere a
trombocitelor, factor mitogen(favotizeaza separatia celulelor),
ADP(agregare).
Placutele
raspund stereotip prin modificari de forma, agregare si
secretia continutului granulelor alfa si a corpusculilor
densi.
La
suprafata placutelor activate se expun receptorii glicoproteici
G.P. II, b/IIIa care fixeaza fibrinogenul. Prin fixarea fibrinogenului pe
doua placute se realizeaza punti proteice, esenta
procesului de agregare.
Pe
suprafata unei placute activate se expun pana la 50000
receptori GP IIb/IIIa care fixeaza fibrinogenul si realizeaza o
structura destul de densa pentru a opri temporar o hemoragie.
La
consolidarea agregatelor contribuie si modificarile de membrana
ce permit patrunderea de macromolecule din plasma in sistemul
canalicular deschis. Ulterior in procesul de coagulare se ajunge la o cimentare
cu filamente de fibrina a agregatelor.
Aderarea
placutelor este procesul de fixare a placutelor pe diferite
suprafete. Depinde de urmatorii factori: natura suprafetei la
care adera; regimul de curgere a
sangelui; prezenta unor proteine cu proprietati adezive; prezenta de glicoproteine receptor in
membrana placutelor.
Placutele
adera la colagen si microfibrilele din peretii vasculari. Au
mare afinitate pentru placi ateromatoase. Acumularea placutelor
la nivelul trombilor se datoreaza atat fixarii de suprafetele
lezate ale vaselor cat si prin incorporarea in reteaua de
fibrina.
Rolul Ca in activitatea placutelor
In
citoplasma placutelor Ca se combina cu o proteina,
calmodulina determinand: activarea fosfolipazei A2 si
amplificarea raspunsului plachetar pe calea producerii de endoperoxizi
si tromboxan A2; activarea unor proteinkinaze C cu rol in
modificarile de forma ale placutelor si in
secretie; activarea unor
fosfodiesteraze care degradeaza c AMP; activarea unor protein fosfataze.
Anomalii cantitative ale
trombocitelor
I.
Trombocitopeniile(scaderea nr. sub 100000/ mm3
)
Clinic:
hemoragii cutanate(petesii si echimoze), digestive, cerebrale; nu se
insotesc de hemoragii articulare sau in tesuturile profunde.
T.S.
prelungit, test al garoului pozitiv - asociere cu fragilitate capilara
si defect de retractie a cheagului.
1). Trombocitopenii prin productie scazuta apare in:
o leucemii, limfoame, tumori metastazante in maduva;
o mielom multiplu, macroglobulinemia Waldenstrom;
o xantomatoze, boli granulomatoase;
o agenti fizici, chimici, biologici ce produc aplazie medulara
ca: radiatii, medicamente, alcool, infectii, diuretice tiazidice
2) Trombocitopenii prin scaderea duratei
de viata
a) Mecanism direct toxic sau mediat
imunologic prin.
o
lezarea placutelor prin
complexe imune;
o
fixarea medicamentului pe
membrana placutelor care devin antigenice si fixeaza anticorpii;
o
formarea de autoanticorpi
fata de componentele antigenice ale placutelor proprii.
Purpura trombocitopenica medicamentoasa
Aproximativ
80 medicamente sunt implicate prin efect mielosupresiv ca: citostatice,
cloramfenicol, fenilbutazona, etanol; actiune toxica
directa: ristocetina, heparina si mecanism imun: sulfamide,
saruri de aur, derivati de chinina, metildopa, AB., antiinflamatoare,
sedative.
Purpura trombocitopenica
idiopatica(PTI) este definita prin: trombocitopenie
variabila, numar normal sau crescut de megacariocite, cresterea
anticorpilor tip IgG fixati pe membrana, lipsa unei alte
afectiuni asociate cu trombopenie, expunere la medicamente sau toxice.
Clinic:
90 % prezinta istoric de infectii virale sau vaccinari
recente; purpura
petesiala, echimoze aparute spontan; bule hemoragice pe mucoasa
bucala; epistaxis; hematurie;
hemoragie digestiva.
Fiziopatologic:
distructia plachetelor de catre macrofage este mediata de Ac IgG
legati de membrana plachetara. Tinta anticorpului este complexul
glicoproteic IIb/IIIa din membrana. Placutele acoperite de
anticorpi sunt distruse de macrofagele splenice. In cazul IgM distructia
se poate face de catre macrofagele hepatice (splenectomia nu este
necesara). Majoritatea prezinta autoanticorpi in plasma si
pe suprafata trombocitelor cauzand scaderea duratei de
viata prin distrugere prematura in splina.
b) Trombocitopenii prin consum exagerat
Apar in: sindroame de C.I.V.D., la nivelul
unor hemangioame gigante; operatii pe cord prin circulatie
extracorporeala; microangiopatii trombotice.
Purpura trombotica
trombocitopenica ( P.T.T.) sau
boala Moschowitz
Mecanismul
este incomplet cunoscut. Se manifesta prin: sindrom hemoragipar cutanat
si mucos; paloare, febra, manifestari neurologice.
3. Trombocitopeniile prin sechestrare in
splina,
4.
Trombocitopenia dilutionala prin aport masiv de lichide.
In
transfuziile masive de sange pe langa efect dilutional poate apare un
defect functional al placutelor, tulburari de coagulare;
dispare la 3 - 4 zile dupa oprirea transfuziilor.
II.
Trombocitozele si trombociteniile(creste numarului aprox.
400000 / mm3 )
A.
Trombocitoza reprezinta cresterea reactiva
a numarului de trombocite.
Apare
in:
o
stari fiziologice: efort
fizic prin mobilizarea trombocitelor din capilarele pulmonare; perioada
ovulatiei; sarcina.
o
stari patologice:
cresterea productiei de placute; interventii
chirurgicale majore (dupa 4 zile); procese inflamatorii acute sau cronice;
cancerul de san, pulmonar(ca sindrom paraneoplazic); anemiile hiperregenerative
(posthemoragice, hemolitice); dupa tratament in anemiile feriprive
cronice; criza reticulocitara dupa tratament cu vit. B12
in anemia Biermer; cardiopatii congenitale cianogene; tumori renale; 50 % din
cei splenectomizati(mecanism necunoscut).
Anomalii calitative
ale placutelor(trombocitopeniile)
I. Anomalii
genetice
Se
suspicioneaza cand avem timp de sangerare prelungit cu nr. plachete normal
sau timp de sangerare (T.S.) foarte lung cu usoara trombopenie.
Trebuie
eliminata o afectare secundara si utilizarea unor medicamente de
obicei apar in copilarie.
Comportamentul
anormal al placutelor poate fi determinat si de factori care nu
tin de placute: modificarile calitative ale structurilor
subendoteliale; anomali calitative ale unor proteine plasmatie a factorului V.
Willebrand si fibrinogenul.
1). Anomalii date de un defect de aderare a
placutelor
a) Sindromul Bernard - Soulier(distrofia trombotica hemoragica)
Evolueaza
cu sindrom hemoragic si se transmite dupa un mecanism autosomal
recisiv, deficit de GPIbIX.
b) Sindrom pseudo - von - Willebrand
Anomalie
transmisa prin mecanism autosomal dominant, evolueaza cu sindrom hemoragic
mucocutanat si cu T.S. prelungit.
c) Deficit de aderare la colagen
Consta
intr-un deficit al glicoproteinei Ia responsabila de interactiunea
placutei cu colagenul.
2. Anomalii determinate de o perturbare a
procesului de activare a placutelor.
3. Anomalii datorate unor defecte cantitative
sau calitative ale corpusculilor densi
4. Deficite functionale ale
placutelor determinate de anomalii ale unor proteine plasmatice
a) B. Von Willebrand (b.v.W.F) prin mecanism autosomal dominant.
Se
datoreaza absentei sau anomaliilor de structura ale unei
glicoproteine plasmatice esentiala pentru functia
placutelor sanguine (f.v.W.F.).Clinic: hemoragii in mucoase,
retroamigdaliene, epistaxis, metroragii.
b) Anomalii ale functiei plachetare in
afibrinogenemie si hipofibrogenemie
Deficitul
total de fibrinogen are un mecanism autosomal recesiv(1 caz la 2 milioane).
Fibrinogenul este necesar procesului de agregare plachetara. T.S. este
prelungit, timpul de agregabilitate este scurt, modificat.
Exista
riscul unor hemoragii cataclismice dupa interventii chirurgicale.
II. Anomali
castigate ale placutelor sanguine
1. Anomalii secundare unor boli si
evoluand cu reducerea functiilor
a)
Dupa medicamente ca: antiinflamatoare nesteroidice, clofibrat,
dipiridamol, heparina, anticalcice (verapamil), blocante ale receptorilor
adrenergici. Riscul este crescut cand se administreaza la hemofilici,
hepatici, uremici, la cei tratati cu anticoagulante orale (anti vit. K).
b)
C.I.V.D. placute sunt astenizate de actiunea endotoxinelor
microbiene, au o reactivitate scazuta.
c)
Uremia; placute hipoagregabile probabil prin retentia
unor substante dializabile (uree, fenoli), hormonul paratiroidian (are
efect inhibitor prin acumularea intraplachetara de c AMP).
d)
Sindroame mieloproliferative(hiporeactivitate plachetara).
f)
Ciroze hepatice si hepatite cronice.
Tulburarea
hemostazei intensive prin anomalii ale coagularii si accelerarii
fibrinolizei. Disfunctiile plachetare survin in legatura cu
excesul de C.I. circulante care se pot fixa pe placute, sau ca urmare
a unor episoade de C.I.V.D.
2. Stari patologice asociate cu
hiperreactivitatea placutelor
sindromul nefrotic (S.N.).
bolile cardio-vasculare.
2.
Hemostaza secundara
Coagularea
propriu zisa reprezinta ansamblul de fenomene care duce la
transformarea fibrinogenului solubil in gel insolubil de fibrina.
Coagularea
are 2 cai:
a)
Calea intrinseca initiala, de activare a f. XII in contact cu
suprafata lezata, se combina cu f. XI rezultand complexul XII -
XI (produsul activarii de contact) care v-a actiona f. IX care se
combina cu f. VIII formand un complex ce activeaza f. X.
Urmeaza
apoi o serie de reactii comune cu calea extrinseca urmate de
eliberarea de mici cantitati de trombina. Trombina (enzima
proteolitica puternica) detaseaza din molecula de
fibrinogen, 2 perechi de fibrinopeptide (A si B) dupa care monomerii
de fibrina ramasi se polimerizeaza formand molecula de
fibrina. Aceasta pentru stabilitate necesita prezenta f. XIII
asigurand soliditatea cheagului.
b)
Calea extrinseca initiata de factorul tisular, tromboplastina
tisulara se combina cu f. VII
si formeaza 'produsul intermediar I' care activeaza in
continuare f. X. Impreuna cu f. V tromboplastina (protrombinaza) care
cliveaza enzimatic o molecula de trombina din molecula din
protrombina. Micile cantitati de trombina au rol de
activare a proceselor anterioare de cresterea debitului caii
intrinseci prin actiuni asupra f. XII si f. V.
Proteaze dependente de vitamina K sunt protrombina (F.II), f. VII,
f. IX si f. X, proteaze sintetizate in ficat in prezenta vit. K.
Vitamina
K are doua forme: naturala din plante si sintetizata de
catre flora intestinala
absorbita in prezenta sarurilor biliare.
Factorul tisular (tromboplastina
tisulara, TF, TPT) este un receptor celular pentru f VII; este un
component al celulelor ce ajunge in contact cu sangele doar in caz de leziuni
tisulare; este o lipoproteina aflata in cantitate mare in endoteliile
vasculare; devine expus pe lumenul vascular dupa stimuli nocivi prin
actiunea IL - 1 si TNF (factor de necroza, tumoare); intima
vasculara din vecinatatea unei placi aterosclerotice este foarte
bogata in TF.
Alte proteaze cu
rol in coagulare sunt: f XII, prekalikreina, f XI si Kininogenul.
Sinteza nu necesita vit. K. Kininogenul este un cofactor al celor trei
proteine, favorizand interactiunea dintre ele prin formarea de complexe.
Activarea prin contact pune in joc atat mecanismul
intrinsec al coagularii cat si fibrinoliza, formarea de kinine
si activarea C.
F XII - Hageman poate
fi activat de catre plasmina, Kalikneina, tripsina.
Luand in considerare faptul ca in plasma nu se
gasesc proteaze active in stare libera, ca exista
inhibitori de proteaze in concentratie mare nu se poate imagina activarea
f XII de catre plasmina sau kalikreina.
Placutele
sanguine au rol in procesul de coagulare pe cale intrinseca astfel:
modificarile suferite de membrana stimulata a ADP-ului confera o
suprafata unde este facilitata activarea f XII.;
Cofactorii coagularii
sunt proteine plasmatice cu rol in coagulare dar lipsite de
proprietati enzimatice.
F
V, f VIII, H M W K G actioneaza prin alinierea proteazei activatoare
(f X) si a zimogenului substrat (protrombina) la suprafata
fosfolipidica a placutelor in pozitii optime pentru interactiune.
Orice leziune vasculara duce la fixarea complexului v.W. f - f VIIIc la
straturile subendoteliale si prin acest complex placutele
adera la aceste structuri.
Formarea si stabilizarea fibrinei
Trombina
actioneaza asupra fibrinogenului si prin scindarea unor
fragmente peptidice(fibronopeptide) il transforma in monomeri de
fibrina care apoi polimerizeaza si formeaza reteaua de
fibrina. Trombina activeaza si f XIII ca 'stabilizator al
fibrinei' care actioneaza ca o transglutaminaza.
Stabileste legaturi covalente intre monomerii de fibrina
polimerizati, conferind stabilitate retelei de fibrina.
Sub
actiunea fXIII se produc legaturi covalente intre fibrina
si fibronectina prin care fibrina se leaga de colagenul
peretelui vascular. F XIII se
gaseste in plasma ca un zimogen inactiv. Este o
transglutaminaza ce stabileste legaturi intre glutamina
si lizina din monomerii de fibrina.
Fibrinogenul,
fibrina, f XIII si fibronectina realizeaza o unitate functionala
cu rol in hemostaza dar si in vindecarea si cicatrizarea
plagilor. F XIII catalizeaza incorporarea alfa2
antiplasminei in reteaua de fibrina crescandu-i rezistenta la
agentii fibrinolitici.
Fibrinogenul este o glicoproteina, se gaseste in
plasma in concentratie mai mare decat alti factori ai coagularii.
Se
gaseste si in granulele alfa ale placutelor, dar,
principala sursa este hepatocitul (scade doar in faza terminala a
afectiunilor hepatice). Fibronectina FN, este o
glicoproteina de adeziune care se gaseste in plasma sub
forma solubila si in forma insolubila legata de
colagen in matricea substantei fundamentalea tesutului conjnctiv.
Intervine
in mentinerea structurii tesuturilor si are actiune
opsonizanta de fagocitare a resturilor celulare, a bacteriilor, fragmentelor
de colagen.
Mecanisme anticoagulante
Depind
de: proprietatile antitrombotice ale endoteliilor; sistemul
fibrinolitic; factori si mecanisme anticoagulante; miscarea
rapida a sangelui prin vene.
Inhibitorii
naturali ai coagularii
1. Antitrombina III (AT III)
Este
o alfa2 glicoproteina plasmatica sintetizata in ficat
si in endoteliile vasculare. A.T.III inhiba: trombina formand un
complex stabil indepartat prin captare in macrofage; f X a si IX a, f
XII a, XI a, kalikreina si plasmina. Efectul creste in prezenta
heparinei.
2. Cofactorul II al heparinei (N.C.II); este inhibitor al trombinei; -
activitatea lui creste in prezenta heparinei.
3. Inhibitorul caii mediale de F.T. (T E
P I )
Este
o proteina plasmatica produsa in E.V. Devine activ dupa
initierea coagularii, inhiba complexul F.T. / f VII a doar
dupa generarea de f X a.
4. Sistemul Proteinei C
Sistem
format din: P.C., proteina S, trombomodulina si actioneaza in
stransa cooperare cu E.V.
Alti agenti anticoagulanti: heparina,
un polizaharid sulfatat sintetizat la nivelul mastocitelor din tesutul
conjunctiv.
Anomalii ale coagularii
I. Anomalii cu caracter genetic
1. Anomalii cu transmitere legata de
sex.
a) Hemofilia clasica (H.A.) este o
deficienta functionala a f VIII manifestata prin
alterarea coagulabilitatii, anomalia genei f. VIII situata pe
bratul lung al crozomului X(defecte in secventa nucleotidelor), de la
simple mutatii punctiforme la deletii importante, in timp ce Ag legat
de complexul f VIII (v.W.F.) este normal sau crescut. Se transmite printr-o
gena patologica legata de cromozomul X. Boala este clinic
manifestata la barbati(femeile sunt purtatoare). Sunt forme
severe, medii, usoare in functie de activitatea f. VIII(1%,1-5%, 5%,
5-25%)
Manifestari:
sangerari spontane in articulatii si muschi, hemartroze,
epistaxis, durere la nivelul sangerarii, hematoame in muschi,
hematurie, sangerari gastro intestinale, sangerari la
extractiile dentare, hematoame retroperitoneale ce explica neuropatia
femurala, sangerare oro faringiana, in SNC(20-30% deces), sunt prinse
articulatiile mari.
Paraclinic: TS normal; TPT crescut; TQ
normal; TC crescut; trombocite normale.
b) Hemofilia B - B. Cristmas
Anomalia
coagularii se datoreaza nu numai lipsei de sinteza, dar mai ales
formarii unor molecule anormale si ineficiente - mai rara.
2. Coagulopatii genetice cu transmitere
autosomala
Afecteaza
egal femeia si barbatul, au mecanism de transmitere recesiv cand
boala apare la homozigoti sau determinant cu grad variabil de
penetranta.
a) Afibrinogenemia congenitala: defect
de sinteza a F transmis autosomal recesiv, homozigotii provin din
casatorii consanguine, se manifesta prin sangerari diverse.
II.
Anomalii cu caracter castigat
Insuficienta
hepatica duce la: scaderea proteazelor serice dependente de vit. K
(Proteina C); scaderea f. V, f
XIII, A.T. III, Proteina S.
Fiziopatologia fibrinolizei
Fibrinoliza
reprezinta degradarea enzimatica a fibrinei de catre
plasmina; este cel mai important mecanism de prevenire a trombozei.
Procesul
fibrinolitic este rezultatul unei interactiuni intre activatorii si
inhibitorii care asigura indepartarea depozitelor endovasculare de
fibrina ce pot duce la tromboza si previn liza prematura a
dopului hemostatic ce s-ar solda cu sangerari prelungite.
Toate
enzimele implicate in fibrinoliza circula in plasma sub
forma inactiva ce se pot activa fie in contact cu fibrina (t-PA) sau
prin proteoliza limitata a plasminogenului. Sunt dotate cu structuri
in ansa capabile de a lega lizina si de a le fixa in reteaua de
fibrina.
Plasmina este substanta care
determina liza cheagului de fibrina. Este generata prin
activarea precursorului inactiv - plasminogenul.
Plasminogenul este o glicoproteina sintetizata in ficat. Forma nativa are ca
aminoacid N - terminal acidul glutamic (Glu - PLG) iar asparagina se afla
in pozitie C-terminala. Activarea plasminogenului de catre t -
PA sau urokinoza (u-PA) implica formarea unei molecule de
plasmina formata din 2
lanturi polipeptidice unite prin 2 punti disulfidice. Plasmina este o
enzima proteolitica similara tripsinei ce contine in centru
activ aminoacizii histadina, aspartat si serina. Este
capabila sa lizeze fibrina dar si fibrogenul, f V si f VII
ai coagularii.
Activatorii fibrinolizei
1. Activatorul tisular al plasminogenului t
- PA
Este
sintetizat in citoplasma celulelor endoteliale, stocat in citoplasma
endoteliilor, eliberat de diversi stimuli ca: hipoxia generala sau
locala(ocluzie venoasa); efort fizic; adrenalina.
2. Urokinaza (activator de tip urinar) U -
PA; descoperit initial de urina; este produs de celule renale, macrofage, celule endoteliale, epiteliile
cailor urinare, cornee, vagin, mucoasa gastrica. Participa
la activarea fibrinolitica bazala si stimulata.
3. Sistem de activare intrinsec; intervin
doar componente ale plasminei.
Alti activatori: kinaze bacteriene;
streptokinaza; stafilokinaza; enzime proteolitice ca: tripsina si papaina.
Inhibitorii
fibrinolizei
1. Inhibitorii activarii
plasminogenului: PAI - 1 - provenit
din endotelii si hepatocit; PAI - 2 - din placenta; PAI - 3 = PCI -
inhibator al proteinei C
PAI
- 1 - sursa in hepatocite, CE, fibroblaste, celule musculare netede,
granule alfa ale placutelor
sanguine. Sinteza este
stimulata de: insulina(cel din hepatocite); IL-1, endotoxina din
endotelii.
PAI-2
in placenta, granulocite, monocite, macrofage, neoplasme ovariene. Are
afinitate mai mare pentru U - PA. Are rol de a proteja organismul matern
fata de o activare brutala a fibrinolizei in sarcina
si expulzie.
PAI
- 3 produs de hepatocite, are efect mai puternic asupra proteinei C
activata.
2. alfa2 antiplasmina, alfa2
plasmin inhibator ( alfa2AP, alfa2PI): are mare afinitate pentru plasmina
blocandu-i centrul activ;
Anomalii ale
fibrinolizei
In
general se asociaza cu anomalii ale coagularii.
I. Anomalii cu caracter familial ale
fibrinolizei
1. Anomalii care evolueaza cu
tendinte de hemoragie: sunt reprezentate de deficite ale inhibitorilor
fibrinolizei; crestere cu caracter familial a productiei de t-PA; se
produce o liza rapida a dopului hemostatic tradusa clinic prin
hemoragii in doi timpi.
2. Anomalii care evolueaza cu
tendinte la tromboze
a) Reducerea activitatii
activatorului plasminogenului: scaderea t-PA in conditii bazale dar
mai ales o lipsa de raspuns in urma stimularii prin ocluzie
venoasa, efort fizic.
b) Anomalii familiale ale plasminogenului: scaderea
concentratiei plasmatice de P. (caracter genetic) - hipoplasminogenemie
familiala; modificari ale
structurii ale moleculei de plasminogen - displasminogemie familiala.
Simdroame fibrinolitice acute
1. Boli medicale:afectiuni
hepatice; sindrom mieloproliferativ: hipervascozitate cu staze si hipoxie
ce favorizeaza eliberarea activatorilor; muscatura de
sarpe: C.I.D. cu activarea plasminogenului prin toxine si enzime;
transfuzii cu sange incompatibil; terapie excesiva fibrinolitica:
exces de streptokinoza si urokinoza; soc electric, termic,
traumatic: hipoxie cu eliberarea activatorilor vasculari fibrinolitici.
2. Boli chirurgicale: interventii pe prostata, uter,
plamani, pancreas: organe bogate in tromboplastina ce induc CID
si-n activatorii tisulari ai plasminogenului ce induc
hiperfibrinoliza; circulatie extracorporeala - activarea f XII.
3. Boli obstetrico - ginecologice.: dezlipire
prematura de placenta: CID tromboplastina; fibrinoliza - activatori placentari;
retentie de fat mort; mola hidatiforma; hematom retroplacentar,
embolie cu lichid amniotic.
Anomalii castigate ale sistemului
fibrinolitic in ciroza hepatica, leucemie
promielocitara, C.I.V.D.;
amiloidoza, sindrom nefrotic etc.
Coagularea intravasculara diseminata
(C.I.D.)
CID
este cea mai complexa anomalie a hemostazei evoluand atat cu tromboze cat
si cu hemoragii. Boli in care poate interveni: septicemie (gram-negativ);
complicatii obstetricale; neoplasme; hemoliza intravasculara;
boli hepatice severe; traumatisme, arsuri; reactii imune intravasculare;
proteze valvulare si vasculare; veninuri si toxine animale.
Din
punct de vedere biochimic CID este o stare de activare anarhica a
sistemelor proteazice (coagulare, fibrinoliza, kininoformatoare).
Principalele
fenomene sunt:
o
formarea de microtrombi
fibrinoplachetari la nivelul capilarelor;
o
consum consecutiv al
placutelor sanguine si ai unor factori ai coagularii;
Clinic: fenomene
trombohemoragice si insuficienta functionala a
organelor cu care s-au produs obstructii trombotice ale
microcirculatiei.
Fiziopatologia C.I.D.
Este
importanta stabilirea secventei fenomenelor biochimice si
fiziopatologice prin intermediul endoteliilor, ale coagularii, sistemului
fibrinolitic si placutelor sanguine.
1. Mecanismele implicate in activarea
coagularii:
Principalul
mecanism in activarea coagularii in C.I.D. este reprezentat de activarea
extrinseca prin tromboplastina tisulara numita T.F. care este
eliberat de C.E., monocite (nu PMN) prin: liza celulelor prin disruptia
membranei; activarea celulei care expune in mod activ F.T. la
suprafata - in special C.E., eliberarea de F.T. de pe suprafata
celulei si trecerea in circulatie impreuna cu fragmente de membrana
(leucemii).
Endotoxinele
pot produce acelasi lucru direct sau indirect pe cai mediate de
macrofage ca produsi de degradare ai C. Expunerea sau eliberarea de F.T.
duce la activarea f VII si coagulare pe cale extrinseca.
Trombina
rezultata potenteaza si calea intrinseca activand f
VIII c cu formarea de fibrina in microcirculatie.
2. Deprimarea activitatii
anticoagulante.
In
C.I.D. se produce deprimarea activitatii anticoagulante prin 2
mecanisme: prin epuizarea diversilor inhibitori prin formarea de complexe
inactive cu factorii activatori ai coagularii; prin leziunile endoteliilor
diminua interventia acestor celule in mecanismele de control ale
coagularii.
Rolul fibrinolizei in C.I.D.
In
prima faza are loc eliberarea de t-PA din endoteliile vasculare si
absorbtia lui pe depozitele de fibrina cu eliberarea de produsi
de degradare ai ei. La aproximativ 3 oare activitatea fibrinolitica din sangele
circulant scade brusc atat prin inactivarea t-PA ca urmare a formarii de
complexe cu PAI cat si datorita epuizarii t-PA circulant in urma
absorbtie pe reteaua de fibrina. Aceasta explica
prezenta produsilor de degradare ai fibrinei in mica circulatie
si lipsa activitatii fibrinolitice.
Persistenta
depozitelor de fibrina se explica prin stimularea mai puternica
procoagulanta a endoteliilor fata de fibrinoliza si
blocarea precoce a fibrinolizei prin exces de inhibitori de tip PAI. Deci
procesul de fibrinoformare il excede pe cel de fibrinoliza.
Rolul placutelor sanguine:eliberarea
de PAi din placutele activate exercita un efect de limitare a
fibrinolizei compensatorii; factorii eliberati din placute ca:
PAF, serotonia, leucotriena LTB4 pot leza endoteliile direct sau
prin radicalii superoxizi produsi de leucocitele stimulate de catre
acesti produsi.
Placutele
activate isi elibereaza continutul, se agrega si sunt
incluse in trombi fibrino-plachetari. Numarul lor scade si cele
ramase sunt hiprreactive.
Scaderea
adezivitatii si agregabilitatii placutelor
ramase in circulatie contribuie la sindromul hemoragic al bolnavilor
cu C.I.D.
C.I.D. s-ar declansa
daca trei procese ar surveni simultan sau consecutiv: activarea coagularii; reactiile vasomotorii; inhibarea
fibrinolizei.
Principalul
mecanism prin care endotoxina declanseaza activarea coagularii
consta in exprimarea de P.T. la suprafata celulelor endoteliale
si a monocitelor.
Intrucat
activarea fibrinolizei este de scurta durata, fiind rapid
blocata de catre inhibitorii de tip PAI, microtrombii vor persista
si blocand microcirculatia duc la necroza si
insuficienta organica.
Pe
de alta parte consumul unor factori ai coagularii si
placutelor; leziunile
peretelui vascular si efectele produsilor de degradare
fibrinolitica creaza conditii unor manifestari hemoragice,
astfel ca la examenul anatomo-patologic gasim leziuni necrotico-
hemoragice.
Clinic se suspicioneaza un episod CID
cand: apare brusc un soc hipotensiv
(cianoza, deces); apare brusc un sindrom hemoragic cu echimoze
generalizate; apar necroze cutanate la extremitati (simetrice); apare
o stare de soc si I.R.A.
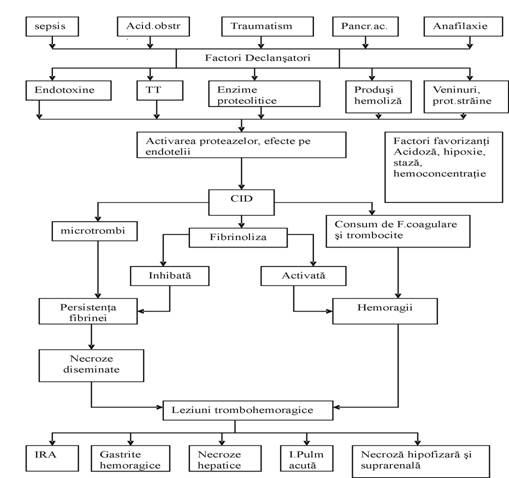
Fig.1. Fiziopatologia
CID

